Tous Pharmaciens La revue n°30 - avril 2026
DOSSIER
Dispositifs médicaux : un rôle stratégique pour les pharmaciens
09/04/2026
Dispositif médical - Exercice professionnel
Omniprésents dans le système de santé, mais parfois méconnus, les dispositifs médicaux (DM) structurent, par leur diversité et leur technicité, une part essentielle du soin. Dans cet environnement complexe, le pharmacien occupe une position stratégique, au cœur des enjeux de sécurité, de traçabilité et de bon usage des DM.
CONTEXTE ET ENJEUX
Présents à différents moments de la prise en charge des patients, les dispositifs médicaux (DM) occupent une place croissante dans les parcours de soins. Selon le code de la santé publique (CSP), un dispositif médical désigne « tout instrument, appareil, équipement, matière, produit […] destiné par le fabricant à être utilisé chez l’homme à des fins médicales, et dont l’action principale voulue n’est pas obtenue par des moyens pharmacologiques ou immunologiques ni par métabolisme, mais dont la fonction peut être assistée par de tels moyens. Constitue également un dispositif médical le logiciel destiné par le fabricant à être utilisé spécifiquement à des fins diagnostiques ou thérapeutiques ». Les DM de diagnostic in vitro (DMDIV), qui répondent à des logiques d’évaluation et de surveillance spécifiques, ne seront pas abordés dans la suite de ce dossier.
Diversité et niveau de risques
Selon la Direction de la recherche, des études, de l’évaluation et des statistiques (DREES), la consommation de dispositifs médicaux en France a représenté 21,7 milliards d’euros en 2024, soit près de 8 % des dépenses de santé en valeur (1) (+ 4,4 % par rapport à l’année précédente). Cette dynamique illustre le poids sanitaire et économique croissant des DM dans le système de santé. Ils couvrent une très large diversité de produits (plus de 20 000, selon l’ANSM [2]), allant des pansements aux implants chirurgicaux, en passant par les logiciels de santé ou encore les masques chirurgicaux. Cette diversité se traduit par une classification en quatre classes de risques croissants :
- la classe I regroupe les dispositifs à risque faible, tels que les pansements ou les béquilles ;
- les classes IIa et IIb correspondent respectivement à des DM avec risques potentiels modérés (lentilles de contact, produits d’obturation dentaire, etc.) et élevés (pompes à perfusion, préservatifs, etc.) ;
- la classe III rassemble les dispositifs critiques, dont la défaillance peut engager le pronostic vital, comme certains implants ou dispositifs de stimulation cardiaque.
Cette classification tient compte de la destination du DM, de sa durée d’utilisation, de la partie du corps exposée (externe ou interne) et des risques pour la santé publique. Aussi, plus la classe du DM est élevée, plus le niveau d’exigence en matière de sécurité sanitaire, de traçabilité et de maîtrise des risques est élevé, domaines dans lesquels le pharmacien occupe un rôle prépondérant.
CADRE RÉGLEMENTAIRE DES DISPOSITIFS MÉDICAUX
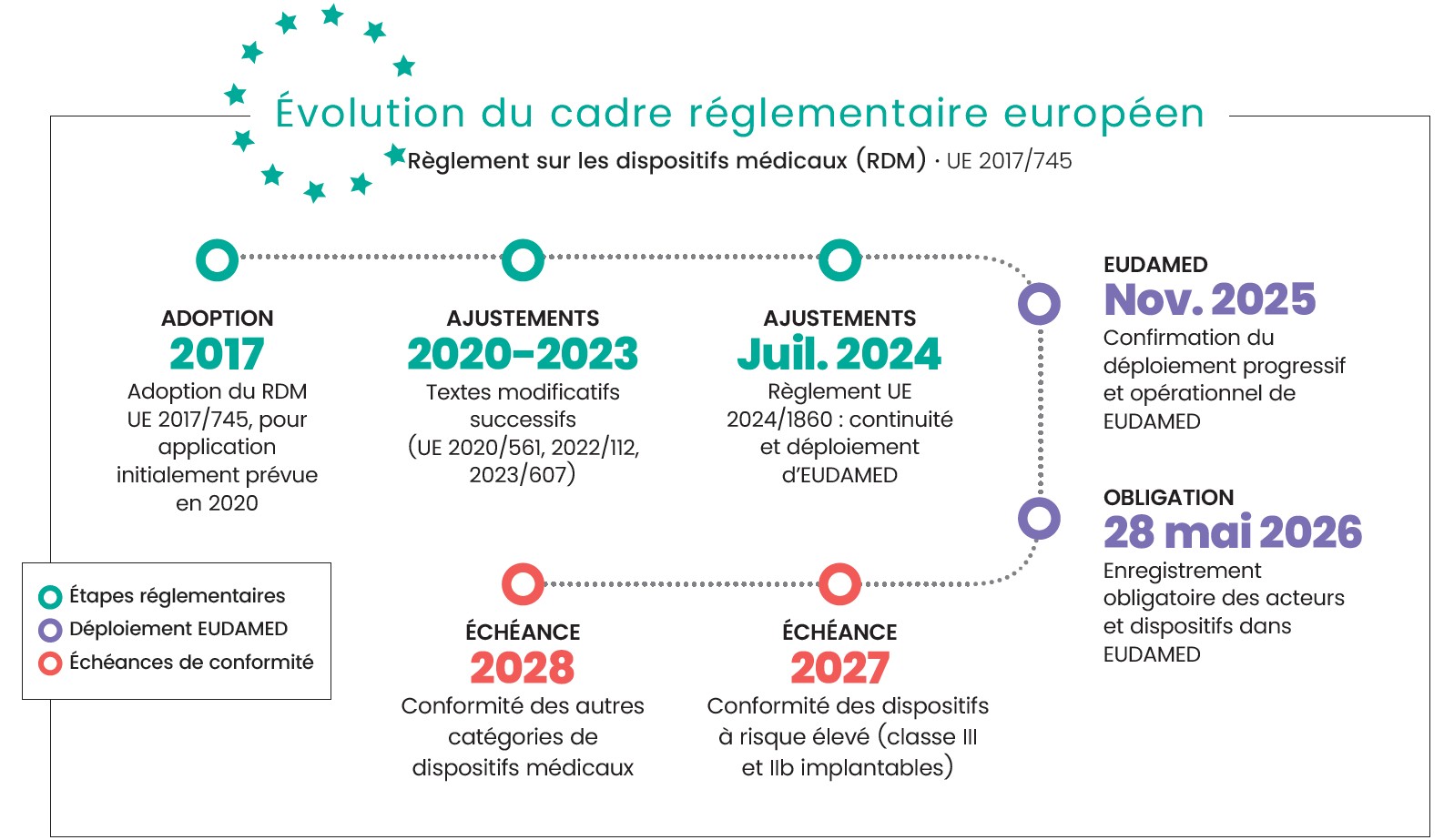
Avant l’adoption du cadre actuel, les DM étaient régis par des directives européennes transposées dans les droits nationaux. L’avènement du règlement européen sur les dispositifs médicaux (RDM) (UE) 2017/745 a permis d’harmoniser progressivement le cadre réglementaire. Ce règlement vise à renforcer la sécurité sanitaire en imposant des exigences accrues en matière de performance, de gestion des risques, d’évaluation clinique et de surveillance du cycle de vie des DM. Il détermine également les règles de classification des DM et les obligations des opérateurs économiques (fabricant, importateur, distributeur). Son cadre a été complété par plusieurs textes modificatifs, en 2020(3), 2022(4) et 2023(5), afin d’échelonner l’application de ses différentes dispositions. Plus récemment, le règlement (UE) 2024/1860, publié en juillet 2024, a introduit une obligation d’information en cas d’interruption ou de cessation d’approvisionnement de certains dispositifs médicaux, tout en accompagnant le progressif de la base de données européenne EUDAMED (6).
À noter que, mi-décembre 2025, la Commission européenne a présenté un projet de règlement visant à simplifier la réglementation des DM et DMDIV, en allégeant notamment certaines procédures d’évaluation, en favorisant l’innovation et l’accès aux dispositifs orphelins et en supprimant la limite de validité des certificats de conformité au profit d’un contrôle périodique. Cette initiative s’inscrit dans une réflexion européenne visant à ajuster le cadre réglementaire, tout en préservant l’accès aux dispositifs médicaux. L’examen du texte pourrait durer plusieurs mois.
EUDAMED
Base de données européenne qui centralise les informations sur les DM commercialisés en Europe, grâce à l’identifiant unique de chaque DM (IUD). Son objectif est de renforcer la traçabilité et la surveillance du marché des dispositifs médicaux à l’échelle européenne, en permettant au public d’avoir accès à des informations sur les dispositifs médicaux commercialisés en Europe, de connaître les incidents déclarés ainsi que l’avancée des investigations cliniques.
Mise sur le marché et conformité
Pour être mis sur le marché, un DM doit être certifié CE. Depuis le 26 mai 2024, un dispositif médical certifié au titre des anciens textes ne peut être mis sur le marché qu’à condition d’avoir fait l’objet, entre autres, d’une demande formelle d’évaluation de la conformité auprès d’un organisme notifié. Pour les dispositifs médicaux de classe I (7), il n’est pas nécessaire de faire appel à un organisme notifié, le processus réglementaire prévoyant une autocertification.
Les organismes notifiés sont des structures indépendantes désignées par les autorités des États membres pour évaluer la conformité des dispositifs.
En France, il en existe deux, le GMED et Afnor Certification. Néanmoins, leur intervention s’inscrit dans un cadre européen : un dispositif fabriqué en France peut être évalué par un organisme notifié établi dans un autre État membre de l’Union européenne.
Afin d’accompagner la transition vers le nouveau cadre réglementaire, une période transitoire a été mise en place.
Plusieurs situations peuvent coexister : dispositifs déjà certifiés conformément au RDM, dispositifs bénéficiant temporairement de mesures transitoires maintenant certaines certifications antérieures, et dispositifs en cours d’évaluation réglementaire. Cette configuration traduit la volonté d’articuler exigences de sécurité renforcées et continuité de prise en charge des patients.
La mise en conformité complète est attendue progressivement, avec un horizon fixé à 2027 pour les dispositifs présentant un risque élevé, notamment les dispositifs de classe III ainsi que certains dispositifs de classe IIb implantables (tels que les vis articulaires) et à 2028 pour les autres catégories de dispositifs.
Par ailleurs, une décision du 27 novembre 2025 a fixé au 28 mai 2026 le délai d’enregistrement obligatoire des acteurs et des dispositifs dans la base de données EUDAMED.
Parole d'expert
Gwennaelle Even
directrice des dispositifs médicaux et des dispositifs de diagnostic in vitro à l’Agence nationale de sécurité du médicament et des produits de santé (ANSM)

Assurer la sécurité
« L’ANSM agit comme une autorité compétente du secteur des dispositifs médicaux et désignante, surveillant le marché français et contrôlant les fabricants pour s’assurer de la conformité et du maintien du bénéfice-risque des dispositifs médicaux. Elle désigne et suit les organismes notifiés français, accompagne l’innovation et autorise les essais cliniques. À travers les inspections et l’analyse des signalements, l’ANSM peut émettre des recommandations, demander des rappels de produits ou prendre des décisions plus contraignantes pour protéger les patients. Elle participe également aux travaux européens afin de garantir un système sûr, opérationnel et un accès continu aux dispositifs innovants et aux populations spécifiques. Elle a par ailleurs travaillé conjointement avec l’Ordre sur des recommandations relatives à la traçabilité des dispositifs médicaux en officine. »
Parole d'expert
Cécile Vaugelade
directrice des affaires techniques et réglementaires au Syndicat national de l’industrie des technologies médicales (Snitem)
Garantir la sécurité du patient
« Le passage au RDM 2017/745 a imposé une remise à plat complète du système, en augmentant tous les prérequis d’exigences de conformité et de démonstration pour obtenir le marquage CE médical. Dans ce cadre, le Snitem joue un rôle de traducteur et de défenseur des intérêts des opérateurs français, notamment en assurant la veille et en décortiquant cette réglementation complexe pour les accompagner dans la continuité des soins et la survie des innovations. L’articulation avec le niveau national repose sur l’ANSM, qui exerce un pouvoir de surveillance du marché et de police sanitaire pour garantir que la libre circulation européenne ne se fasse jamais au détriment de la sécurité des patients en France. »
LA CHAÎNE DU DISPOSITIF MÉDICAL : RESPONSABILITÉS ET EXIGENCES À CHAQUE ÉTAPE
La commercialisation d’un DM implique des acteurs multiples : fabricants, mandataires, importateurs, distributeurs, établissements de santé, professionnels de santé… Les pharmaciens, en fonction de leur exercice, exercent des responsabilités variées dans la gestion de ces dispositifs, bien qu’ils ne relèvent pas tous du monopole pharmaceutique.
Fabrication et mise sur le marché
La fabrication et la commercialisation des DM sont assurées par les fabricants. À la différence du médicament, ces deux activités ne sont pas obligatoirement gérées par des pharmaciens. Les fabricants sont responsables de la conformité réglementaire du dispositif avant sa mise sur le marché. Cela inclut notamment l’obtention du marquage CE, grâce aux organismes notifiés, pour attester de la conformité aux exigences de sécurité et de performance, la constitution et la conservation de la documentation technique, ainsi que la gestion des analyses de risques et des données cliniques lorsque cela est requis.
En France, la surveillance du marché des DM relève de l’ANSM, sauf pour les dispositifs médicaux dits « grand public », pour lesquels elle partage cette compétence avec la DGCCRF (8).
Dans le cadre de ses missions de surveillance du marché, l’ANSM s’assure que les dispositifs disponibles en France sont sûrs d’utilisation. À ce titre, elle réalise entre autres la centralisation des déclarations d’incidents, rend des avis sur la qualification et classification des DM, mène les inspections des sites fabricants, contrôle la conformité des dispositifs mis sur le marché. Elle est également l’autorité compétente pour leur publicité. Elle participe aussi à la construction de la réglementation européenne relative aux DM.
Distribution et circulation des dispositifs médicaux
La distribution assure la continuité entre la production et l’utilisation. Sont considérés comme distributeurs tous les acteurs mettant un DM à disposition sur le marché, tels les grossistes-répartiteurs, les dépositaires, mais aussi les pharmaciens d’officine (au sens du règlement UE sur les DM).
À ce titre, ils doivent garantir la traçabilité des DM, le respect des conditions de stockage et de transport, la gestion des retraits et rappels de lots : « Nous devons vérifier le certificat CE, la déclaration de conformité, la validité du certificat, la présence du marquage CE avec le numéro de l’organisme notifié, l’adresse du fabricant, la notice, le cas échéant, la présence des mentions obligatoires et les pictogrammes réglementaires, par exemple sur les DM stériles », explique Marie-France Bertrand, chargée de mission coordination collection à la Compagnie d’exploitation et de répartition pharmaceutique (CERP). Pour les pharmaciens d’officine – à la fois professionnels de santé et distributeurs –, l’ANSM a récemment mis en ligne un guide rappelant leurs obligations en tant que distributeurs (à retrouver sur le site de l’ANSM, rubrique Actualités).
Identification des opérateurs et traçabilité des dispositifs médicaux : une responsabilité partagée tout au long de la chaîne
Le règlement (UE) 2017/745 a renforcé la traçabilité des dispositifs médicaux, avec la mise en place de l’identifiant unique du dispositif (IUD), qui permet de reconnaître chaque produit (fabricant, lot ou série, dates), d’assurer sa lecture standardisée tout au long de la chaîne logistique, d’améliorer la gestion des alertes sanitaires et des rappels de lots, et d’enregistrer ces données dans la base EUDAMED. Cet IUD repose sur deux composantes :
- un identifiant de modèle (IUD-DI), fixe pour un même dispositif, qui permet son identification et son enregistrement dans la base européenne EUDAMED ;
- un identifiant de production (IUD-PI) qui regroupe des données variables (numéro de lot, numéro de série, dates) et permet d’assurer la traçabilité opérationnelle.
La traçabilité recouvre par ailleurs deux dimensions complémentaires :
- la traçabilité des flux entre opérateurs économiques imposant aux fabricants, importateurs et distributeurs d’identifier leurs fournisseurs et leurs clients professionnels, et de conserver ces informations pendant au moins dix ans (quinze ans pour les dispositifs implantables) ;
- la traçabilité des dispositifs médicaux eux-mêmes, obligatoire pour les dispositifs les plus à risque, notamment les dispositifs médicaux implantables de classe III, pour lesquels les identifiants doivent être enregistrés et conservés par les acteurs de la chaîne, ainsi que par les établissements de santé.
La coexistence de formats multiples (codes barres, Datamatrix, structures d’encodage variables selon les pays ou les industriels) « complique l’intégration automatique des données dans les systèmes logistiques, renforçant les enjeux d’interopérabilité des outils de traçabilité », souligne Jean Brevilliers, président du Conseil central de la section C, représentant les pharmaciens de la distribution en gros, directeur des affaires pharmaceutiques et pharmacien responsable au sein d’un réseau de grossistes-répartiteurs.
Pour les pharmaciens, cette exigence se traduit par la capacité à vérifier la conformité des produits à retracer les flux et à garantir la fiabilité des informations tout au long du circuit.
Témoignage
Paul Beyou
directeur des affaires pharmaceutiques au sein d’un laboratoire pharmaceutique, conseiller ordinal, membre du bureau du Conseil central de la section B, représentant les pharmaciens de l’industrie
La règle des produits combinés
"Le statut d’un produit combiné – DM et médicament – dépend de l’action revendiquée par l’ensemble : si l’action pharmacologique domine, le statut médicament l’emporte ; si la substance n’a qu’un rôle accessoire et que l’action est mécanique ou physique, c’est le statut dispositif médical qui prévaut. Par exemple, un stent coronarien enrobé d’une substance active est classé comme dispositif médical, car sa fonction principale reste le maintien mécanique de l’ouverture vasculaire, la substance associée n’ayant qu’un rôle complémentaire. Dans ce cadre, l’organisme notifié évalue la conformité du dispositif et sollicite un avis rendu par l’Agence nationale de sécurité du médicament et des produits de santé."
Délivrance, utilisation et suivi
Cette étape engage directement la responsabilité professionnelle du pharmacien. Il doit en effet assurer l’information du patient ou du professionnel utilisateur sur les conditions d’usage du DM, la traçabilité selon les dispositifs délivrés, la sécurité d’utilisation, incluant la vérification de la compatibilité du dispositif avec la situation clinique et les conditions de conservation. Ces obligations participent à la gestion du risque sanitaire, lié aux dispositifs médicaux.
- Les DM non stériles relèvent d’une surveillance standard et nécessitent un contrôle des conditions de conservation et d’usage, notamment pour prévenir toute altération fonctionnelle du DM.
- Les DM stériles présentent, quant à eux, des exigences renforcées : les pharmacies à usage intérieur (PUI) ont pour mission d’assurer la gestion exclusive des DM stériles dans les établissements où elles sont situées. L’enjeu principal est de maintenir la stérilité du DM stérile jusqu’à son utilisation finale. Cela suppose la préservation de l’intégrité du protecteur individuel de stérilité, une manipulation limitée et contrôlée, un stockage respectant les conditions environnementales recommandées et une vigilance particulière sur les dates de péremption.
Témoignage
Michaël Peres
pharmacien biologiste médical, CHRU de Nancy, conseiller ordinal section G, représentant les pharmaciens biologistes médicaux
Le dispositif médical au service du diagnostic
"Dans la pratique du biologiste médical, l’utilisation des dispositifs médicaux est très limitée par rapport aux DMDIV. Leur classification dépend de leur utilisation finale. Ils constituent avant tout des outils au service du diagnostic (tubes, seringues ou écouvillons), notamment au cours de la phase pré-analytique du prélèvement, dont la qualité et la maîtrise conditionnent directement la fiabilité des résultats analytiques."
Témoignage
Grégory Pape
pharmacien d’officine dans les Yvelines, conseiller ordinal section A, représentant les pharmaciens titulaires d’officine
Accompagner l’usage du DM à l’officine
"À l’officine, le pharmacien joue un rôle d’intermédiaire et d’accompagnateur dans le champ très large des dispositifs médicaux. Il s’assure d’abord que le dispositif correspond au besoin réel du patient, dans le cadre fixé par la prescription, puis il traduit l’information technique en conseils pratiques pour permettre une utilisation correcte et sécurisée du produit. Ainsi, qu’il s’agisse d’un matériel de mobilité, d’un pansement ou d’un dispositif de mesure, le pharmacien agit comme un relais entre le fabricant, le prescripteur et le patient."
Parole d'expert
Isabelle Le Du
présidente de la Société pharmaceutique française des dispositifs médicaux (SPFDM) – Europharmat
Le pharmacien, acteur clé du dispositif médical
"Le domaine du dispositif médical est extrêmement vaste et diversifié : le pharmacien doit disposer d’une expertise réglementaire, technique, mais aussi d’une maîtrise du bon usage et des règles de référencement au sein des établissements de santé. Cette polyvalence le positionne comme un acteur central de la chaîne du dispositif médical, à l’interface des praticiens, des patients et des enjeux économiques. Dans un contexte d’évolution réglementaire et d’essor, notamment dernièrement des DM numériques, la formation constitue un levier stratégique. La SPFDM s’inscrit dans cette dynamique, en structurant les compétences des deuxièmes et troisièmes cycles de la formation des pharmaciens et en accompagnant les professionnels tout au long de leur carrière."
Les dispositifs implantables, quelles spécificités ?
Le dispositif médical implantable (DMI) est défini comme « tout dispositif […] destiné à être totalement introduit dans le corps humain ou à remplacer une surface anatomique ou une fonction, par une intervention clinique, et à y rester après la procédure. Un dispositif partiellement implanté destiné à rester au moins 30 jours est également considéré comme implantable »*.
Les DMI sont utilisés dans plusieurs spécialités cliniques : cardiovasculaire (stents, valves, pacemakers), orthopédique (prothèses articulaires), neurologique (neurostimulateurs, implants cochléaires), oto-rhino-laryngologique ou encore dentaire et maxillo-faciale.
On distingue les DMI actifs, nécessitant une source d’énergie pour fonctionner (stimulateurs cardiaques, défibrillateurs implantables, pompes à insuline) ou non actifs (prothèses articulaires ou mammaires, stents).
Tous les DMI, sauf exception, sont classés IIb ou III, correspondant aux dispositifs à risque élevé.
Pour les DMI de classe III, la traçabilité est renforcée : tous les opérateurs économiques doivent enregistrer et conserver l’identifiant unique des dispositifs (IUD) qu’ils ont fournis ou qu’on leur a fournis. En France, cette exigence est étendue à l’ensemble des DMI, à l’exception de certains dispositifs (sutures, vis, agrafes…), pour lesquels la traçabilité n’est pas obligatoire.
Les établissements de santé, notamment grâce à leurs PUI, sont aussi soumis à des obligations de traçabilité des dispositifs médicaux implantables, qui doit être assurée à chaque étape jusqu’à l’implantation au patient.
Quel cadre régit les DMI ?
Le RDM UE 2017/745 a été complété par des dispositions françaises visant la traçabilité et l’information du patient, dont l’arrêté du 8 septembre 2021 relatif au management de la qualité du circuit des DMI. Il impose aux établissements de mettre en œuvre un système de management de la qualité couvrant l’ensemble du circuit des DMI, d’assurer la traçabilité sanitaire à chaque étape et de délivrer au patient une information complète sur le DMI qui lui a été implanté.
* Définition du RDM UE 2017/745.
Témoignage
Gilbert Bounaud-Devillers
patient porteur d’un défibrillateur, chargé de mission à l’Association de porteurs de dispositifs électriques cardiaques (Apodec) et membre d’un groupe de travail sur les DMI à France Assos Santé
Une information des patients encore perfectible
"Malgré l’évolution réglementaire, les patients repartent chez eux avec une information incomplète, parfois avec des cartes d’implants rendues difficilement lisibles, voire rédigées en langue étrangère, ce qui rend l’accessibilité à l’information assez opaque pour les patients. Cette mesure reste perfectible : si le cadre normatif organise formellement les obligations des établissements en matière d’information, de traçabilité et de remise de documents au patient, son effectivité opérationnelle demeure dépendante des modalités organisationnelles locales, de la qualité des supports d’information effectivement remis et de l’implication des professionnels de santé dans la transmission compréhensible des données."
VIGILANCE ET SÉCURITÉ
Les dispositifs médicaux requièrent une attention particulière tout au long de leur cycle de vie. Leur utilisation s’inscrit dans un cadre de gestion des risques visant à prévenir, détecter et maîtriser les événements susceptibles d’altérer la qualité des soins ou la sécurité des patients.
Surveillance post-commercialisation
La sécurité sanitaire des dispositifs médicaux repose sur un système global de surveillance et de vigilance couvrant l’ensemble du cycle de vie du produit, depuis sa conception jusqu’à son utilisation en conditions réelles. Contrairement aux médicaments, la mise sur le marché des DM ne nécessite pas forcément la réalisation d’études cliniques. Par conséquent, la surveillance post-commercialisation constitue le principal mécanisme, permettant d’évaluer de manière continue le rapport bénéfice-risque du DM, à travers la collecte de données sur sa qualité, ses performances et sa sécurité.
Cette surveillance permanente permet, le cas échéant, de prendre les mesures préventives ou correctives appropriées.
Matériovigilance : sécuriser l’utilisation des DM
La matériovigilance a pour objet la surveillance des incidents ou des risques d’incidents résultant de l’utilisation des DM. Elle s’applique à tous les DM, indépendamment de leur classe ou de leur état de stérilité. Elle permet de détecter, d’analyser et de prévenir les incidents liés à l’utilisation des dispositifs médicaux, tels que les dysfonctionnements ou altérations de leurs performances, les erreurs d’utilisation, le défaut d’informations fournies par le fabricant, susceptible d’affecter la sécurité des patients, ou encore les effets indésirables. La matériovigilance s’inscrit dans un processus continu de signalement, d’analyse et de mise en œuvre de mesures correctives adaptées, pouvant aller jusqu’au retrait du marché, lorsque la situation l’exige.
Ce système repose sur un circuit de signalement associant l’ensemble des acteurs du parcours de soins et de la chaîne industrielle, ainsi que les patients.
Les professionnels de santé, notamment les pharmaciens exerçant en établissement de santé, contribuent au signalement et à la qualité des informations transmises, tandis que les fabricants sont responsables de l’analyse des incidents et de la mise en œuvre des actions correctives. Bien que cela soit moins fréquent, les pharmaciens d’officine sont aussi concernés.
En savoir plus :
Parole d'expert
Yves Lurton
Yves Lurton, pharmacien praticien hospitalier au CHU de Rennes, correspondant local de matériovigilance et coordonnateur régional de matériovigilance et réactovigilance Bretagne*
Instruction et filtrage des alertes
"Le correspondant local de matériovigilance assure une double mission d’instruction et de filtrage des données de matériovigilance de son établissement. Il instruit les signalements issus des services de soins (alertes ascendantes) et les transmet aux fournisseurs ainsi qu’à l’ANSM, si nécessaire. Concernant les alertes descendantes, il effectue une veille quotidienne sur le site de l’ANSM pour identifier, parmi les alertes nationales, celles qui concernent directement les produits référencés et utilisés dans son établissement. Depuis 2022, un échelon régional vient renforcer le circuit de matériovigilance en France, assurant un lien entre les correspondants locaux et l’ANSM afin d’optimiser le traitement des signalements."
Signalement des incidents : quelles obligations ?
Les pharmaciens ayant connaissance d’un incident grave ont l’obligation, comme tout professionnel de santé, de le déclarer à l’ANSM (ou auprès de leur correspondant local de matériovigilance lorsqu’ils exercent en établissement de santé). Ils peuvent déclarer, en outre, tous les autres incidents dont ils ont connaissance, suspectés d’être dus à un dispositif, auprès du fabricant afin que celui-ci puisse exercer ses activités de surveillance après commercialisation.
Un incident est qualifié de grave lorsqu’il a entraîné directement ou indirectement, ou est susceptible d’avoir entraîné ou d’entraîner :
- la mort d’un patient, d’un utilisateur ou de toute autre personne ;
- une grave dégradation, temporaire ou permanente, de l’état de santé d’un patient, d’un utilisateur ou de toute autre personne ;
- une menace grave pour la santé publique.
La qualité des informations déclaratives est un facteur déterminant pour l’analyse technique des incidents. L’identification précise du dispositif concerné – référence produit, numéro de lot ou numéro de série –, la conservation du DM en lui-même, la description des circonstances de survenue de l’incident, les conséquences cliniques observées, sont autant d’informations nécessaires qui contribuent à la réalisation d’une expertise appropriée et à la mise en place de mesures correctives.
Toute personne peut également effectuer un signalement direct de matériovigilance, sur le portail de signalement des événements sanitaires indésirables seul ou avec l’aide d’un professionnel de santé, comme le pharmacien.
Outils d’information et d’alerte
Les fabricants et distributeurs ont une obligation d’informer leurs clients en cas de mesure de sécurité, rappel ou retrait de lots de DM. Ces alertes doivent être mises en œuvre sans délai par les distributeurs, établissements de santé, pharmaciens. Une base d’alerte nationale, placée sous l’autorité de l’ANSM, recense les dispositifs médicaux rappelés, les signalements de matériovigilance ainsi que les recommandations de sécurité associées. Le site de la DGCCRF, Rappel Conso, héberge quant à lui les rappels/retraits de DM dits « grand public » (exemple : béquille). Le système du DP-Rappels, applicable pour le médicament, ne permet pas de diffuser des rappels/retraits de DM.
Il est recommandé au pharmacien de s’abonner aux alertes de l’ANSM et de la DGCCRF pour recevoir les mesures de sécurité mises en place pour les DM.
À cet égard, l’utilisation de standards d’identification lisibles automatiquement, tels que les codes barres ou les identifiants bidimensionnels de type Datamatrix, et plus largement la traçabilité assurée par l’IUD, permet la reconnaissance rapide des produits concernés par une alerte et facilite la mise en œuvre des retraits ciblés, renforçant ainsi l’efficacité de l’ensemble du dispositif de vigilance.
Le pharmacien comme vigie
En résumé, la contribution du pharmacien à la sécurité des dispositifs médicaux s’inscrit dans une approche globale de gestion des risques sanitaires. À chaque étape du circuit, il intervient à un niveau spécifique, prenant part à la sécurisation des produits, à leur bon usage et à la détection des incidents.
En amont, il participe à la sécurisation de la chaîne logistique grâce au contrôle de la conformité des produits distribués, au respect des conditions de conservation lors du stockage et du transport, ainsi que grâce à des approvisionnements appropriés. Il peut également contribuer, notamment en industrie, à la surveillance post-commercialisation et à l’évaluation du rapport bénéfice-risque des dispositifs, même si sa présence dépend de l’organisation des entreprises, le secteur ne relevant pas d’un monopole pharmaceutique.
Au moment de la délivrance et de l’utilisation, le pharmacien joue un rôle dans le contrôle de la conformité des produits (en qualité de distributeur au sens du règlement UE 2017/745), l’accompagnement des patients, la diffusion des informations lors des rappels ainsi que la déclaration des incidents d’usage. Il participe aussi, pour certains dispositifs, à la traçabilité en assurant l’enregistrement et la transmission des informations nécessaires à leur suivi.
Les pharmaciens hospitaliers jouent un rôle central dans la sécurité des DM au sein des établissements de santé. Ils assurent la traçabilité des DMI ainsi que la stérilisation des DM réutilisables. Ils contribuent également, en collaboration avec les autres acteurs techniques de l’établissement – ingénieurs biomédicaux, services d’achat, directions des systèmes d’information pour les dispositifs intégrant une composante logicielle –, à la réception et l’analyse des signalements d’incidents issus des services de soins. Les pharmaciens hospitaliers spécialisés en matériovigilance prennent en charge la gestion et diffusion des alertes sanitaires, qu’elles soient ascendantes ou descendantes.
Plus largement, l’ensemble des pharmaciens, qu’ils exercent en officine, en pharmacie à usage intérieur ou dans la distribution, sont des acteurs à part entière de la matériovigilance et du dispositif de signalement, en tant que professionnels de santé de proximité en mesure d’identifier, de recueillir et de transmettre tout incident lié à l’utilisation d’un DM.
Parole d'expert
Marie-France Bertrand
pharmacienne et chargée de mission coordination collection à la Compagnie d’exploitation et de répartition pharmaceutique (CERP)
"Nous assurons une traçabilité rigoureuse"
"Notre métier de grossiste-répartiteur va bien au-delà de la simple logistique : nous agissons comme un véritable filtre de sécurité, en validant systématiquement la conformité réglementaire et documentaire de chaque dispositif médical avant son stockage. Nous assurons une traçabilité rigoureuse et un approvisionnement quotidien des officines, garantissant que seuls des produits certifiés parviennent aux patients. En tant qu’experts du référencement, nous restons un maillon essentiel de la matériovigilance, prêts à signaler aux autorités la moindre anomalie ou effet indésirable dont nous aurions connaissance."
UNE NÉCESSAIRE TRANSITION ÉCOLOGIQUE
La transition écologique liée aux DM est un enjeu de santé publique. Selon The Shift Project, les DM génèrent environ 7,4 millions de tonnes équivalent CO2 par an, soit 20 % des émissions du secteur. L’augmentation des volumes, le plastique et le suremballage accentuent le gaspillage, notamment à l’hôpital et à l’officine où des boîtes surdimensionnées restent partiellement utilisées.
Concilier développement durable et sécurité
Le cadre réglementaire évolue : par exemple, le décret du 19 mars 2025 encadre la remise en bon état d’usage de certains DM, et l’arrêté du 13 mars 2025 limite la délivrance des articles pour pansements par le pharmacien à sept jours, pour éviter surstocks et pertes.
Par ailleurs, une expérimentation nationale, issue de la loi de financement de la Sécurité sociale (LFSS) 2024, instaure (appel à candidatures lancé en février 2026) le retraitement de certains DM à usage unique (par exemple, en cardiologie interventionnelle) dans un cadre sécurisé. Cette démarche, déjà pratiquée en Allemagne, concilie sécurité et réduction de l’empreinte environnementale et s’inscrit dans le RDM, permettant le retraitement par opérateurs certifiés.
Parole d'expert
Pr Valérie Sautou
pharmacienne hospitalo-universitaire, directrice de la transformation écologique au CHU de Clermont-Ferrand
Une évolution nécessaire
« Les dispositifs médicaux ont un impact environnemental majeur, de leur fabrication à leur élimination. En tant que pharmaciens, nous ne pouvons plus ignorer cette réalité. La sécurité des patients reste non négociable, mais considérer l’impact environnemental dans nos décisions doit désormais être aussi naturel que d’intégrer le critère économique. La première question à se poser est simple : avons-nous réellement besoin de tout ce que nous consommons ? Repenser les pratiques, comparer usage unique et réutilisable lorsque c’est possible, introduire des critères environnementaux dans les achats… Ce n’est pas un retour en arrière, c’est une évolution nécessaire. Si nous n’anticipons pas ces enjeux aujourd’hui, nous les subirons demain. Le pharmacien a un rôle clé pour accompagner cette transition, à l’interface entre soins, achats et réglementation. »
Mieux choisir les DM en établissement de santé
Les DM peuvent être à usage unique, ou réutilisables et restérilisables. Le choix entre ces deux options doit notamment prendre en compte leur impact environnemental et les conditions de leur utilisation. L’évaluation du besoin est cruciale : volumes adaptés, instruments utilisés, alternatives réutilisables. L’analyse de cycle de vie guide les décisions, complétée par l’optimisation des emballages, le tri et le recyclage. Des initiatives émergent, comme le Green Bloc pour des blocs opératoires sobres ou la labellisation de maternités. De plus, les achats publics intègrent désormais des critères environnementaux via le Schéma de promotion des achats publics socialement et écologiquement responsables (Spaser) et l’index DM durable (impact climatique, ressources, recyclabilité, dimensions sociales), se dirigeant progressivement vers des alternatives plus vertueuses.
(1) Panorama de la DREES – Les dépenses de santé 2024.
(2) Agence nationale de sécurité du médicament et des produits de santé.
(3) Règlement (UE) 2020/561.
(4) Règlement (UE) 2022/112.
(5) Règlement (UE) 2023/607.
(6) European Database on Medical Devices.
(7) Hors DM de classe I mis sur le marché à l’état stérile, ayant une fonction de mesurage ou les instruments chirurgicaux réutilisables.
(8) Direction générale de la concurrence, de la consommation et de la répression des fraudes.
MOT D'ORDRE
Carine Wolf-Thal,
présidente du Conseil national de l’Ordre des pharmaciens

Les dispositifs médicaux occupent une place croissante dans les prises en charge, et engagent pleinement la responsabilité professionnelle des pharmaciens, quel que soit leur mode d’exercice. Dans un cadre réglementaire européen exigeant, complexe et en constante évolution, leur maîtrise ne peut pas être approximative. L’Ordre national des pharmaciens rappelle que l’exercice de cette responsabilité suppose une connaissance actualisée des textes, une vigilance constante et un engagement dans la formation continue.
À l’heure où les dispositifs médicaux intègrent désormais des composantes numériques, algorithmiques et d’intelligence artificielle, le pharmacien doit plus que jamais affirmer son rôle de garant de la sécurité, du bon usage et de la protection des patients.